16 de mayo: Día Mundial del Angioedema Hereditario
La enfermedad mortal de la que nadie habla y pocos conocen
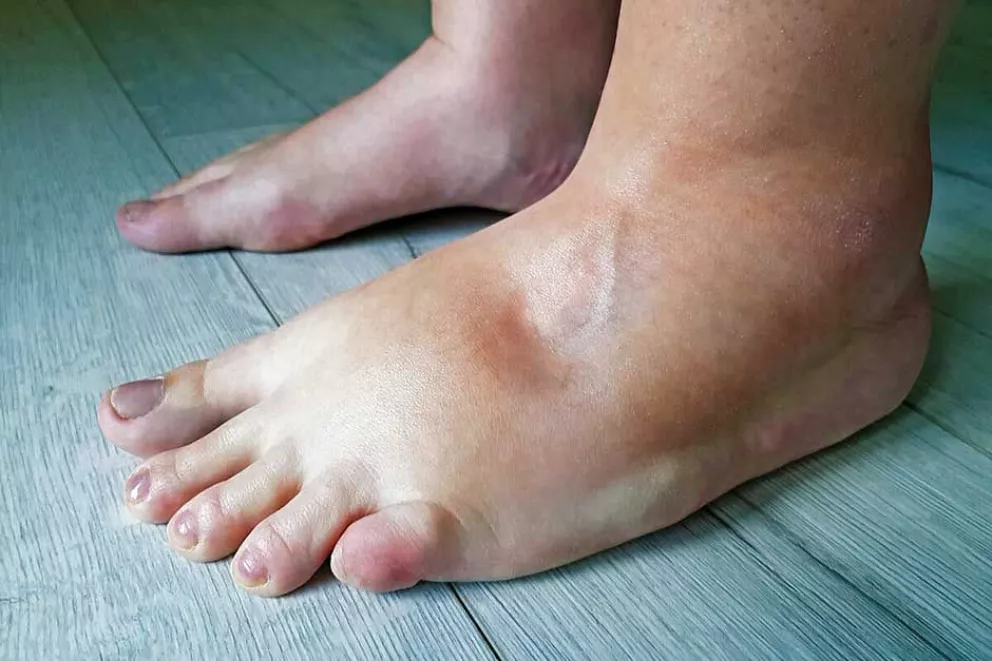
Probablemente a la mayoría de las personas las palabras angioedema hereditario (AEH) no le suenen a nada, pero ¿qué es? Se trata de una enfermedad poco frecuente, pero potencialmente mortal que se caracteriza por episodios recurrentes de hinchazón dolorosa en cara, lengua, glotis (laringe), manos, pies, genitales o abdomen, que duran de dos a cinco días.
No se acompañan de ronchas ni picazón y no mejoran con corticoides, antihistamínicos ni adrenalina y también se la confunde con un cuadro alérgico, pero no lo es.
No siempre hay antecedentes dentro de una familia, aunque si lo hubiera, hace altamente sospechosa la patología.
Para que el AEH se pueda diagnosticar y tratar adecuadamente es necesario conocerlo, tanto por trabajadores de la salud como por pacientes.
Cuando se manifiesta en la glotis puede provocar muerte por asfixia. Cuando se manifiesta en el abdomen simula un cuadro de abdomen agudo: dolor abdominal intenso, vómitos diarrea y distensión abdominal marcada (parece un embarazo de tan hinchado que se pone el abdomen). Muchos pacientes son operados innecesariamente.
Se produce por la liberación masiva de un mediador llamado bradiquinina, responsable de los síntomas. En las variantes más frecuentes, la alteración está en una proteína plasmática llamada C 1 Inhibidor (puede estar en valores bajos, o normales pero no ser funcional). Pero cada vez más se descubren nuevas variantes con C1 Inhibidor normal, donde el defecto genético está en los genes de otras proteínas plasmáticas (Factor XII de la coagulación, kininógeno, angiopoyetina, etcétera).
Todas se manifiestan de manera similar y requieren la misma terapéutica específica. En Misiones tenemos pacientes con y sin defectos del C1 inhibidor que, con frecuencia, afectan a familias enteras, por su característica hereditaria. La mayoría demora muchos años en obtener el diagnóstico correcto.
Marcelo Dante Strass
Especialista en Alergia e Inmunología
Sanatorio Boratti
Posadas, Misiones
Miembro del Comité Científico de AEH de la Asociación Argentina de Alergia e Inmunología (AAAeIC)
En detalle
Tratamiento
El tratamiento de las crisis consiste en administrar medicamentos específicos cono concentrado de C1 inhibidor, que repone los niveles de esta proteína, o una droga llamada Icatibant, que bloquea el receptor de la bradiquinina.
Anticuerpo monoclonal
Últimamente se aprobó en Argentina la utilización de un anticuerpo monoclonal llamado Lanadelumab que bloquea al precursor de la bradiquinina, o sea la kalicreína y se utiliza para profilaxis a largo plazo, logrando que el paciente prácticamente no tenga síntomas.
Aumentan casos
A diferencia de lo que sucede en otras provincias, en Misiones hay más pacientes con AEH con C1 INH normal (defectos del gen del Factor XII): 17 pacientes, que el AEH tipo I: 12 pacientes, haciendo un total hasta la fecha de 29 pacientes. Afortunadamente no todos con síntomas.
Una atención temprana puede salvar vidas
El inicio del angioedema hereditario suele ocurrir durante la infancia o adolescencia, aunque algunos pacientes comienzan sus síntomas en la adultez. La demora promedio en obtener el diagnóstico correcto en Misiones es de 15 años, pero existen pacientes que los han presentado por hasta 30 años antes de saber cuál era la patología que padecían.
Los desencadenantes de las crisis pueden ser estrés, traumatismos, enfermedades infecciosas (vacunarse contra la gripe es recomendable), estrógenos (embarazo, anticonceptivos, pre-menstruo), cirugías o procedimientos odontológicos o que afecten vías aéreas superiores (intubación, endoscopías). El diagnóstico del angioedema debido a alteraciones del C1 inhibidor se realiza midiendo en sangre el C4, el C1 Inhibidor cuantitativo y su funcionalidad. En las formas con C1 inhibidor normal, el diagnóstico es por análisis genético.
En Argentina hay dos medicamentos para las crisis agudas: concentrado de C1 inhibidor (derivado plasmático de donantes) o una droga llamada Icatibant, que bloquea el receptor de la bradiquinina. Ambas se pueden utilizar en niños y adultos. Cuando no se dispone de ninguna de ellas, se sugiere plasma fresco congelado, como tercera opción. El tratamiento debe ser lo más precoz posible para que sea eficaz, idealmente, antes de una hora del comienzo de los síntomas.
Antes de cirugías se utiliza el concentrado de C1 inhibidor como profilaxis a corto plazo. Si bien hay drogas antiguas como los andrógenos atenuados, presentan muchos efectos colaterales y hoy la esperanza en Argentina para el tratamiento a largo plazo está puesto en el lanadelumab, un anticuerpo monoclonal, de reciente aprobación por Anmat.